Cell Growth . Metabolism . Heart Failure . Receptor Tyrosine Kinase . Neuregulin . mTOR
Cardiobiology
Regulation of cardiac cell growth – implications for heart disease
Our group investigates mechanisms whereby receptor tyrosine kinases regulate hypertrophic and hyperplastic cell growth in the heart. To this end we use primary neonatal rat heart cultures and several in vivo mouse models of cardiac disease. During the past reporting period we have investigated models of high-fat diet-induced cardiomyopathy and aortic-constriction-induced cardiac hypertrophy to mimic the pathophysiology associated with obesity and valve disease, respectively. In addition, we are establishing a model of anthracycline-induced cardiomyopathy, relevant to the interdisciplinary field of oncology and cardiology. Our focus at the cellular level is on protein turnover mechanisms, which to a large extent depend on available energy resources together with hormone and cytokine levels. We have recently directed our attention to the ErbB family of receptor tyrosine kinases activated by neuregulin-1β (NRG-1), while also extending our earlier work on the in-sulin/IGF-I/Akt/mTOR pathway.
Neuregulin-1β
NRG-1 regulates cell growth and differentiation in various tissues including the heart. After myocardial infarct or anthracycline therapy, NRG-1 is released by the endothelium and activates multiple signaling pathways leading to distinct biological effects depending on the expression levels and dimerization of its receptors ErbB2, ErbB3 or ErbB4 on neighboring cells in the heart. NRG-1 is cardioprotective in a range of animal models of cardiac disease. While its therapeutic use is being tested in heart failure, the hormone risks to enhance tumor growth. A better understanding of the mechanisms whereby NRG-1 acts in cardiac versus cancer cells is important. We reported that NRG-1 causes glucose uptake in neonatal cardiomyocytes by triggering translocation of GLUT4 to the plasma membrane via ErbB2/ErbB4 (Fig. 1). Using Seahorse, we showed that NRG-1 also enhances glycolysis. Like for insu- lin and IGF-I, the mechanism involves PI3Kα, Akt and AS160 (Pentassuglia et al., 2016; Heim et al., 2020). Under stress conditions cardiomyocytes switch from fatty acids to glucose as energy source. We hypothesized that the NRG-induced glucose uptake contributes to cardiomyocyte contractility e.g. under ischemic conditions or in insulin-resistant states. However, our experiments demonstrated that NRG-1 does not activate this cardiac glucose uptake pathway in adult models. We are currently testing whether the metabolic effects of NRG-1 provide the means for cell cycle activation and proliferation observed in neonatal cardiomyocytes (Fig. 2). While our studies reveal mechanisms that contribute to normal physiological cardiac growth and differentiation observed in neonatal hearts, they also aim to provide fundamental insights that may contribute to cardiac regenerative approaches.
Function of mTORC2 in the heart
To perform its function as biological pump that provides oxygen and nutrients to our whole body, the heart consumes large amounts of energy. A tight regulation of the available resources, including cellular proteins, becomes critical in disease states where metabolism must increase to maintain cardiac performance, e. g. in hypertensive or valve disease. We previously reported that the metabolic regulator mTOR is essential for cardiac function when part of mTORC1: its cardiomyocyte-specific ablation causes heart failure rapidly followed by death of the mice, a phenotype explained by mTORC1’s role in protein synthesis as well as mitochondrial metabolism. We next demonstrated that aortic constriction-induced pressure overload significantly increases rictor (an essential mTORC2-specific protein) and PKCβII and PKCδ phosphorylation in control mice, but not in cardiac rictor knockout mice. Whereas pressure-overload causes hypertrophy with maintained function in controls, it leads to systolic dysfunction in rictor-deficient hearts without having any effects on cardiac weight or fibrosis. These data suggest that mTORC2 regulates metabolism and contractility of the heart via PKCβII and PKCδ and is not implicated in its hypertrophic growth (Shende et al., 2016; Xu et al., 2016).
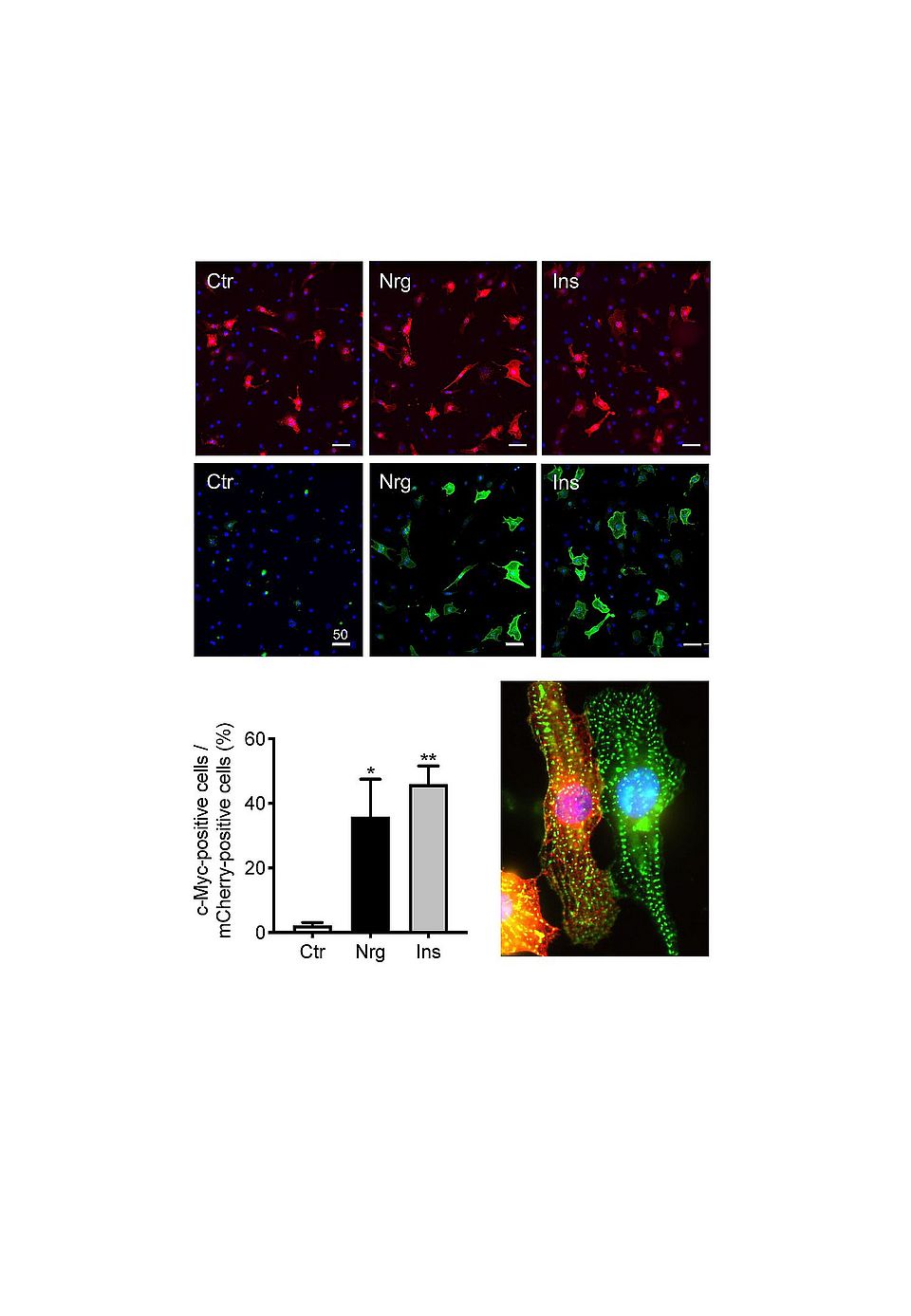
Fig. 1: NRG-1 causes translocation of GLUT4 to the plasma membrane. pLenti-myc-GLUT4-mCherry transfected cardiomyocytes from neonatal rats were stimulated with NRG-1 (Nrg, 10 ng/ mL), insulin (Ins, 13 nM) or vehicle (Ctr) and fixed with 4 % paraformaldehyde after 30 min. The GLUT4-transfected cells are revealed based on the mCherry label (red, top pictures). Translocated GLUT4 was detected on the surface of the fixed non-permeabilized cells using a c-Myc-specific antibody followed by an Alexa 633-labeled secondary antibody (green). The bottom picture identifies the left mCherry-positive cells (red) as cardiomyocytes after staining with antibodies specific for sarcomeric actinin (green), The nuclei were stained with DAPI (blue).
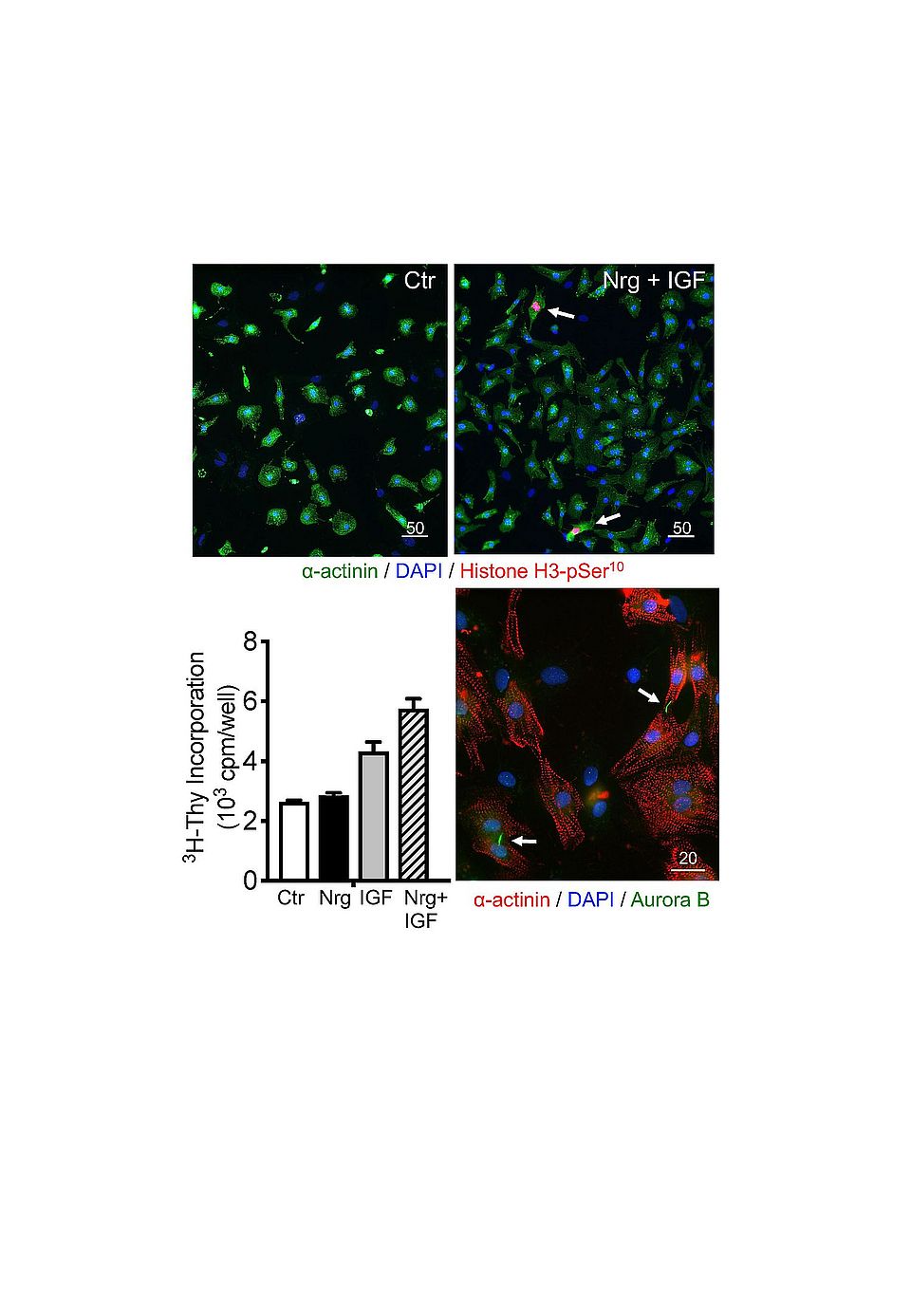
Fig. 2: Effects of IGF-I and NRG-1 on cardiomyocyte cell cycle activation. Cardiomyocyte cultures prepared from neonatal rats were stimulated with NRG-1, IGF-I or both factors together for 24h. Cell cycle activation was evaluated by measuring 3H-thymidine incorporation and by immunofluorescent labeling with antibodies to phosphorylated Histone H3 (top). To confirm cytokinesis in cardiomyocytes, double labelling with antibodies to α-actinin and Aurora B was performed (Bottom micrograph). Bar size indicated in µm.