Neuropsychiatric Disorders . Congenital Developmental Disorders . Hereditary Cancer Syndromes
Human Genomics
Human Genomics: investigating the molecular basis of human genetic disesases
The research group “Human Genomics” aims to identify the molecular (genetic) basis of human diseases and the underlying molecular mechanisms and pathomechanisms by combining human genetics knowledge with new genomics technologies (project leader: Per Hoffmann), bioinformatics and biostatistical approaches and in-depth phenotyping. We focus on genetically complex neuropsychiatric disorders (project leader: Sven Cichon, DMB’s Focal Area Neurobiology), congenital developmental disorders (project leader: Isabel Filges), and hereditary cancer syndromes (project leader: Karl Heinimann, DBM’s Focal Area Oncology). We closely interact with the research group Skin Biology and have a strong interest in the molecular basis of genodermatoses (project leader: Bettina Burger) and hereditary angioedema (project leader: Sven Cichon).
Neuropsychiatric disorders
One strong focus of the research group is the genetic analysis of neuropsychiatric disorders, in particular bipolar disorder. In the context of the Psychiatric Genomics Consortium (PGC), we performed the so far largest genome-wide association study (GWAS) including 20,352 cases and 31,358 controls of European descent (Stahl et al., 2019). 30 loci were genome-wide significant, including 20 newly identified loci (Fig. 1). The significant loci contain genes encoding ion channels, neurotransmitter transporters and synaptic components. Pathway analysis revealed nine significantly enriched gene sets, including regulation of insulin secretion and endocannabinoid signaling. The study also showed that the clinical subgroup Bipolar I disorder is strongly genetically correlated with schizophrenia, driven by psychosis, whereas bipolar II disorder is more strongly correlated with major depressive disorder. These findings address key clinical questions and provide potential biological mechanisms for bipolar disorder.
In parallel we performed exome sequencing studies in large and multiply affected bipolar disorder families (e. g. Maaser et al., 2018). Current and future projects aim at a mechanistic understanding of common and rare genetic risk variants for bipolar disorder. This will include sequencing in further large families with bipolar disorder as well as the study of sets of common risk variants in induced pluripotent stem cell (iPSC) models.
Congenital developmental disorders
Our specific interest relates to birth defects and multiple congenital anomaly syndromes which present early during pregnancy, since the monogenic aetiology of most severe fetal anomalies is poorly understood. In addition, the clinical description of most genetic conditions relates to paediatric and adult patients, and we are trying to understand how those same conditions may present in the prenatal period.
We systematically investigate families with one or more fetuses presenting with distinctive anomaly patterns and identified novel disease genes, new fetal phenotypes and phenotypes as a variable of developmental timing (Meier et al., 2017; Meier et al., 2019; Meier et al., 2020). We further characterized the function of KIF14 in humans and zebrafish (Reilly et al., 2019) and reviewed the emerging role of kinesin family member genes in birth defects, which we proposed to term “kinesinopathies” as a recognizable entity (Kalantari&Filges, 2020; Fig. 2).
Our group also studies Arthrogryposis multiplex congenita (AMC), a heterogeneous group of conditions with multiple contractures (Filges et al., 2019). In an international consortium we work towards a standardized interdisciplinary approach to diagnose and care for patients with AMC.
Hereditary colorectal cancer syndromes
In a collaborative study on autosomal dominant Juvenile Polyposis Syndrome (JPS), we analysed data on almost 700 JPS patients. Compared with BMPR1A carriers, SMAD4 carriers displayed anaemia twice as often, exclusively showed overlap symptoms with haemorrhagic telangiectasia and an increased prevalence of gastric juvenile polyps. Cancer, reported in 15 % of JPS patients, mainly occurred in the colorectum and the stomach. Our results facilitate recommendations for clinical management, and contribute to SMAD4 and BMPR1A databases (Blatter et al., 2020).
In addition, we collated prospective clinical data on 80 Lynch syndrome patients harbouring pathogenic DNA mismatch repair (MMR) gene variants. Integration into the Prospective Lynch Syndrome Database (PLSD) resulted in studies assessing cancer incidence, prognosis, gene-specific cancer risks and the effect of current surveillance measures on mortality (Seppala et al., 2019; Dominguez-Valentin et al., 2020). Research collaborations on RET germline alterations in osteosarcoma patients and on colorectal carcinogenesis, particularly MMR deficient cancers, were successfully published (Kovac et al., 2020; Cross et al., 2018).
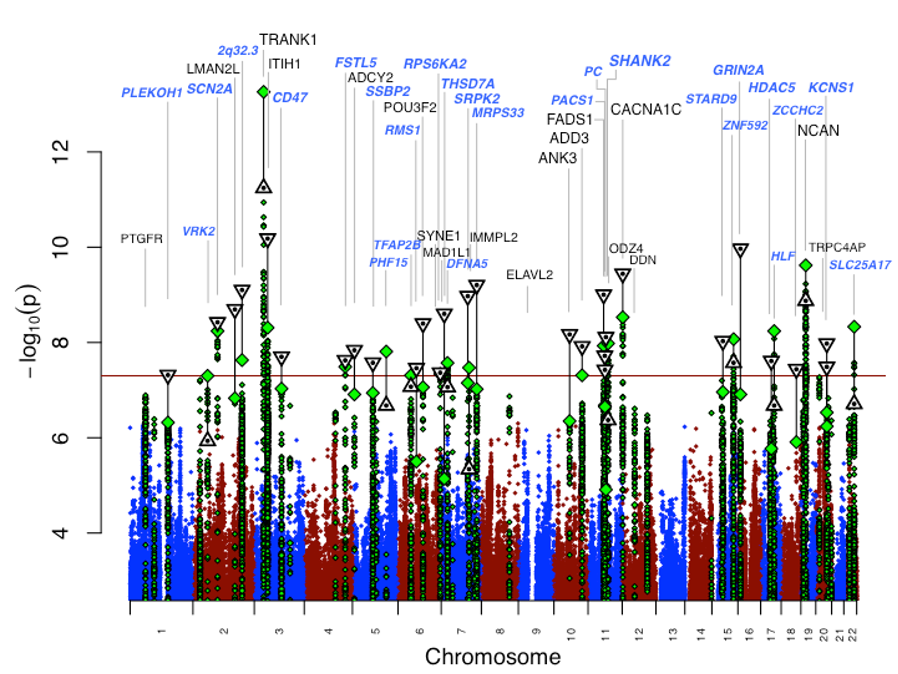
Fig. 1: Results of a large GWAS of bipolar disorder (Stahl et al., 2019, Nat. Genet.): Manhattan plot for our primary genomewide association analysis of 20,352 cases and 31,358 controls. GWAS -log10P-values are plotted for all SNPs across chromosomes 1-22 (diamonds, green for loci with lead SNP GWAS P < 10-6). Combined GWAS+followup -log10Pvalues for lead SNPs reaching genome-wide significance in either GWAS or combined analysis (triangles, inverted if GWAS+followup -log10P > GWAS -log10P). Labels correspond to gene symbols previously reported for published loci for bipolar disorder (black) and the nearest genes for novel loci (blue), at top if GWAS+followup P < 5x10-8.
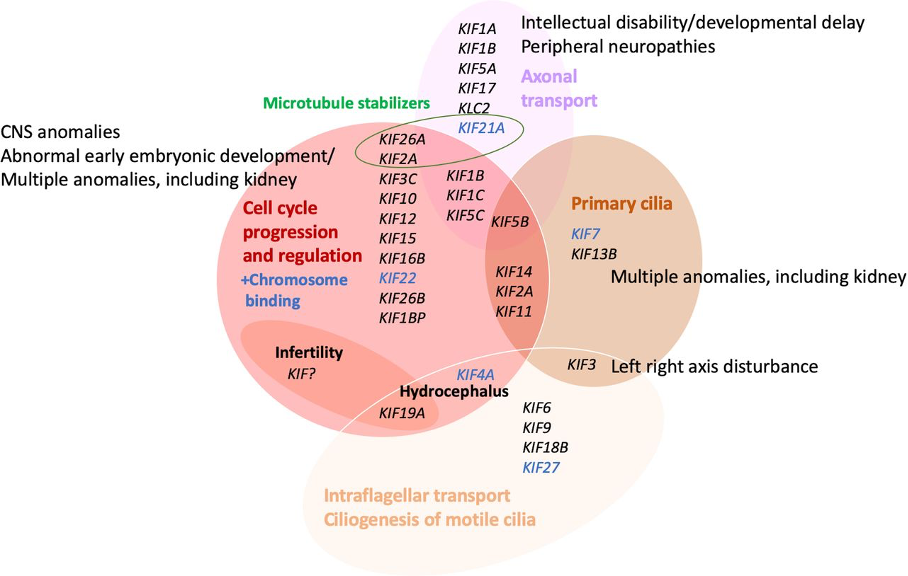
Fig. 2: Congenital developmental disorders: Assignment and clustering of KIF genes to various functions and relation to birth defect or monogenic phenotype groups. Detailed phenotypes are shown in tables 1 and 3. Cancer and multifactorial conditions are not included. CNS, central nervous system (from Kalantari, S. & Filges, I. (2020) J. Med. Genetics).