T cells . B cells . Cellular Metabolism . Organismal Metabolism . Immune Dysregulation
Immunobiology
Immunometabolic regulation of adaptive immunity in health and disease
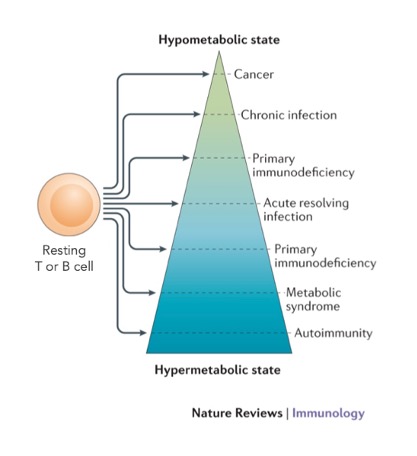
Our research is focused on basic and translational aspects of lymphocyte function and its metabolic basis. Conceptually, our recent work has led us to propose the idea that a spectrum of immune cell metabolic states can provide a basis for categorizing diseases (Fig. 1).
In that scheme, acute resolving infection represents properly regulated cellular metabolism. Exploring this notion experimentally, we tested how acetate – a short chain fatty acid – is regulating T cell metabolism during resolving infection in mice. We found that acetate transiently increases in the blood circulation upon acute infection. This leads to acetylation of the enzyme GAPDH in memory CD8+ T cells, which enhances their glycolytic switch and interlinked inflammatory capacity. At sites of prolonged inflammation, by contrast, acetate accumulates but now catalyzes glutaminase activity and develops suppressive capacity by buffering calcium (Balmer et al. Immunity 2016 and Cell Metabolism 2020). These findings support the idea of an orchestrated early hypermetabolic-, followed by a hypometabolic T cell state jointly enabling resolution of acute infection.
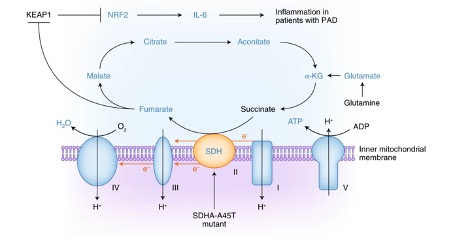
Two disease categories were investigated to further probe our metabolism-centric categorization scheme (Fig. 1), namely cancer and primary antibody deficiency (PAD). In cancer patients, we investigated how tumor-derived TGF-β suppresses the key antitumor function of CD4+ T cells, IFN-γ production. Suppression required expression and phosphorylation of Smad proteins in the TGF-β signaling pathway – but not their nuclear translocation, and it depended on oxygen availability, suggesting a metabolic basis for these effects. Indeed, TGF-β substantial-ly impaired the ATP-coupled respiration of CD4+ T cells and specifically inhibited mitochondrial complex V (ATP synthase) activity. Inhibition of ATP synthase alone was sufficient to impair IFN-γ production by CD4+ T cells. These results suggest that TGF-β targets T cell metabolism directly, diminishing T cell function by driving a hypometabolic phenotype (Dimeloe et al. Science Signaling 2019). In PAD, we previously found that loss-of-function mutations can lead to immunodeficiency by causing a hypometabolic T cell phenotype (Kolev et al., Immunity 2015). In the current reporting period we built on this observation and prospectively screened glycolysis and mitochondrial respiration in B cells from patients with PAD. The highest oxygen consumption rate values were detected in three study participants with persistent polyclonal B cell lymphocytosis (PPBL). Exome sequencing identified germline mutations in SDHA, which encodes succinate dehydrogenase subunit A, in all three patients with PPBL. SDHA gain-of-function led to accumulation of fumarate in PPBL B cells, which engaged the KEAP1-Nrf2 system to drive the transcription of genes encoding inflammatory cytokines. In a single patient trial, blocking the activity of the cytokine interleukin-6 in vivo prevented systemic inflammation and ameliorated clinical disease. Overall, this study thus identified a hypermetabolic phenotype, driving pathological mitochondrial retrograde signal-ing, as a disease modifier in PAD (Burgener et al. Nat. Imm., 2019) (Fig. 2).
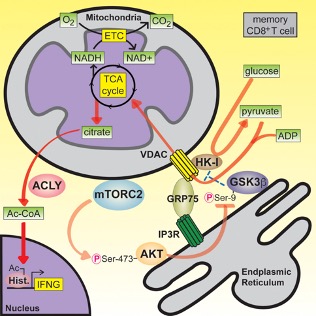
In our basic research efforts we aimed to understand, at the molecular level, how glycolysis is linked to the rapid response of memory CD8+ T cells (Gubser et al., Nat. Imm., 2013). We found that rapid activation of AKT by mTORC2 leads to inhibition of GSK3β at mitochondria- endoplasmic reticulum (ER) junctions. This enabled recruitment of hexokinase I (HK-I) to the voltage-dependent anion channel (VDAC) on mitochondria. Binding of HK-I to VDAC promoted respiration by facilitating metabolite flux into mitochondria. Glucose tracing pinpointed pyruvate oxidation in mitochondria, which was the metabolic requirement for rapid generation of IFN-γ in memory T cells. Subcellular organization of mTORC2-AKT-GSK3β at mitochondria-ER contact sites, promoting HK-I recruitment to VDAC, thus underpins the metabolic reprogramming needed for memory CD8+ T cells to rapidly acquire effector function (Bantug et al. Immunity 2018), (Fig. 3).