Virus . Transplantation . HIV . Immunodeficiency . Acute Infection . Chronic Infection
Transplantation and Clinical Virology (TCV)
Missing immunity and Viral Diseases
The research group “Transplantation and Clinical Virology” focuses on translational aspects of clinically relevant virus infections in humans. Our approach is includes contrasting acute versus chronic viral infections with respect to pathogenesis and immune control using community-acquired respiratory viruses (CARVs) versus persisting human herpesviruses or human polyomaviruses in immunologically naïve, in immunodeficient, or in immunosuppressed patients.
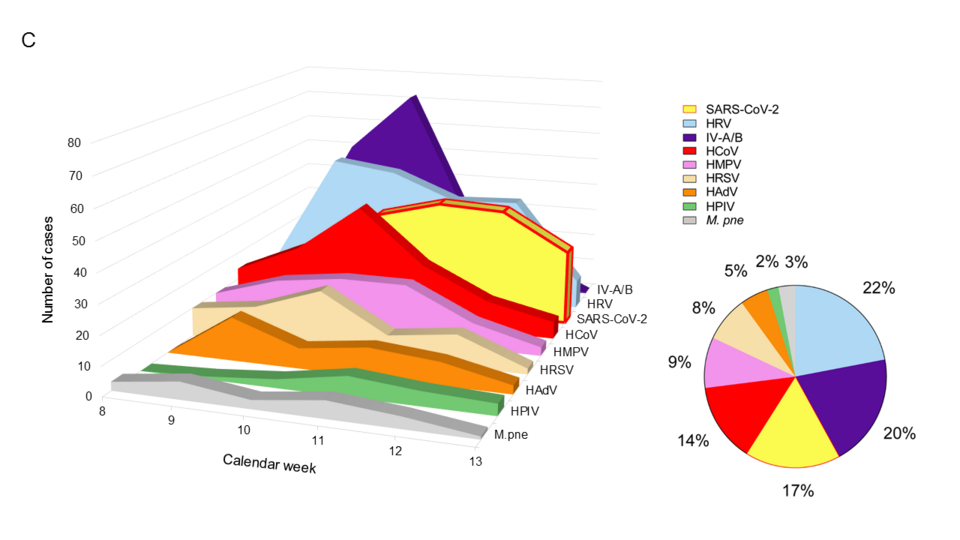
Regarding CARVs, we have been actively engaged in several studies characterizing the epidemiology and impact in solid organ transplantation (SOT) patients participating in the Swiss Transplant Cohort Study (STCS) and in hematopoietic stem cell transplant (HSCT) patients. We contributed to prospective studies of lung transplant recipients and to evaluating a novel antiviral drug (presatovir) targeting fusion of respiratory syncytial virus in HSCT patients. We reviewed the available literature and contributed to CARV- and CoVID19-guidelines in the framework of ECIL and EHA. The year 2020 has been dominated by the SARS-CoV-2 pandemic for which we established a robust in-house assay to diagnose patients and identified its rapid replacing all commonly circulating CARVs (Leuzinger et al. 2020; Fig.1).
Regarding human herpesviruses, cytomegalovirus (CMV), herpes simplex virus (HSV) and Epstein-Barr virus (EBV) are the most relevant challenges. Epidemiology and impact have been evaluated in collaborations with the STCS and locally with Profs Schaub/Steiger. We established phenotypic acyclovir resistance testing for HSV, genotypic resistance for HSV and CMV including the new drug (letermovir). We established CMV-DNAemia as the major form of plasma CMV loads resulting from naked unprotected viral genome fragments. This explains the long-known difficulty of CMV culture from blood as opposed to broncho-alveolar lavage fluids despite high CMV-DNA loads. Our observations have pathophysiological, diagnostic, and therapeutic relevance, and entered the international TTS consensus guidelines on CMV management in SOT.
Regarding human polyomaviruses, we focus on BK polyomavirus (BKPyV) and characterized its epidemiology and impact in collaborations nationally with the STCS, locally with Profs Schaub/Steiger in kidney transplant patients as well as internationally, with the pediatric kidney and HSCT programs in Helsinki, Finland. We improved BKPyV-detection establishing that BKPyV-DNAemia is the major form of “viremia” in transplant patients similar to CMV. These observations impact concepts regarding the use of neutralizing antibodies for protection and therapy. Together with Prof Parmjeet Randhawa, Pittsburgh (PA, USA) and AST-IDCOP, we provided the 2019 clinical guideline on BKPyV in kidney transplantation.
Our experimental virology studies focus on BKPyV in primary human renal tubular epithelial cells as key target of BKPyV nephropathy in kidney transplants. For the first time, we identified an evolutionarily conserved functional role of viral agno- protein, which promotes innate immune evasion by disrupting the mitochondrial membrane potential, fragmenting the mitochondrial network, and targeting the damage mitochondria for autophagy (Manzetti et al. 2020; Fig. 2).
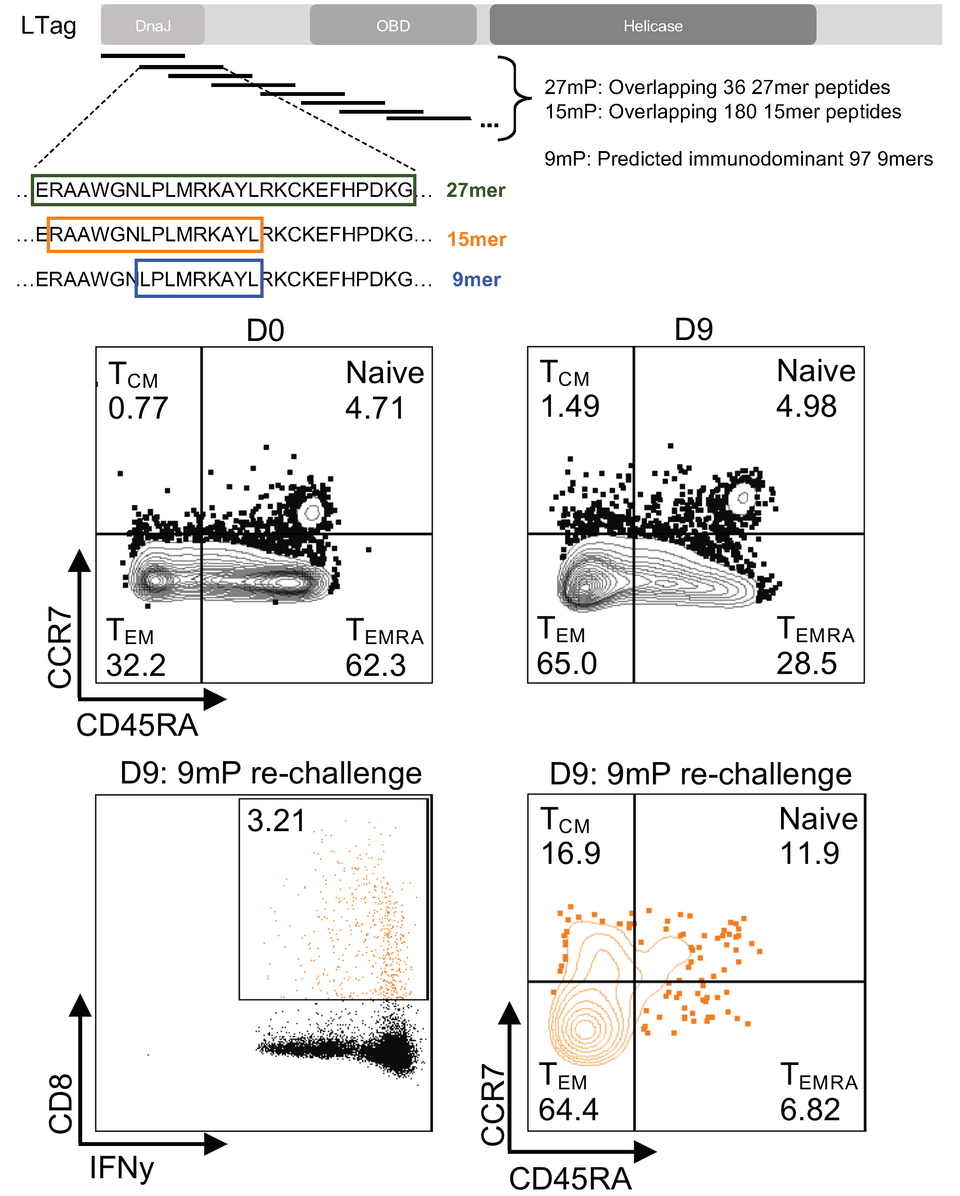
Regarding adaptive immunity, we identified more than 70 immunodominant 9merepitopes recognized by BPyV-specific CD8 T-cells controlling BKPyV-DNAemia. Notably, we developed a novel peptide expansion protocol of BPyV-specific CD8 T-cells in vitro en route to a safe peptide-based protection by adoptive T-cell transfer and vaccination (Wilhelm et al. 2020; Fig. 3). We also identified variant amino acid exchanges in the conserved BKPyV-early protein, which mediate escape from CD8 T-cell control (Leuzinger et al. 2020 10.3390/v12121476).
We plan to reconstitute 3-dimensional renal tubule kidney culture and organoid models for BKPyV infection and antiviral T-cell control as an animal-free surrogate for pre-clinical models of viral immune control and to design phase I clinical vaccination studies in healthy volunteers.
Connection to Clinical Practice
Prof. Manuel Battegay, Prof. Nina Khanna Infectious Diseases & Hospital Epidemiology, University Hospital Basel
Prof. Michael Tamm, Prof. Daiana Stolz Clinic of Pneumology and Pulmonary Cell Research, University Hospital Basel
Prof. Jürg Steiger, Prof. Stefan Schaub, Clinic for Transplantation Immunology and Nephrology, University Hospital Basel
Prof. Mike Recher Immunodeficiency Laboratory, Department of Biomedicine, University and University Hospital of Basel
Prof. Dr. Dr. Adrian Egli Applied Microbiology Research, Laboratory Medicine, and Clinical Bacteriology and My
Liaison dangereuse – Virus infection and missing immunity
“Transplantation and Clinical Virology” is interested in translational research of acute versus persistent virus infections to dissect the pathogenesis of viral diseases, to improve diagnostic tools for clinical use, and to identify strategies of prevention and treatment. Key viral threats are being addressed according to their importance as defined by frequency, severity, and available medical treatment. Thus, we are interested in
– Community-acquired respiratory virus (CARV) infections in populations without sufficient immune control including SARS-CoV-2;
– Emerging epidemics including vector-borne viral diseases (tick-borne encephalitis virus, dengue, Chikungunya, West Nile virus, Zika virus)
– Viruses affecting immunocompromised patients (HIV/AIDS; solid organ transplantation; hematopoietic cell transplantation; autoimmune diseases; primary/inherited immunodeficiency disorders).
Acute virus infections are exemplified by CARVs, while persistent virus infections are represented by human herpes- and polyomaviruses. Both, acute and chronic virus infections are naturally widespread in the general immunocompetent population, but may take a severe course in patients without functional immune memory either because of primary infection, primary immunodeficiency, acquired immunodeficiency or immunosuppression.
We aim at characterizing: 1) key determinants of virus pathology; 2) potential targets of antiviral intervention; 3) relevant innate immune mechanisms; 4) protective targets of adaptive immune memory; 5) modifiable and non-modifiable risk factors in patients to tailor and optimize interventions.