Myeloproliferative Neoplasms . Myeloid Malignancies . JAK2 Tyrosine Kinase . JAK2 Signaling . MAPK Signaling . Kinase Inhibitors . Therapeutic Targeting . Resistance Mechanisms
Myeloid Malignancies
Mechanisms and targeting of oncogenic signaling in myeloproliferative neoplasms
Myeloproliferative neoplasms (MPN) are chronic leukemias with dysregulated production of mature myeloid cells. MPN occur as essential thrombocythemia with thrombocytosis, polycythemia vera with erythrocytosis, or myelofibrosis with increased megakaryocytes and marrow fibrosis. They progress to bone marrow failure or acute myeloid leukemia. Hematopoietic stem cell transplantation as a curative therapy is limited to a subset of patients. It is the goal of our studies to contribute to novel therapeutic approaches by targeting the molecular signaling driving these diseases.
MPN are characterized by dysregulated signaling of the JAK2 kinase due to acquired mutations in the JAK2 signaling pathway. JAK2 is a tyrosine kinase essential for hematopoiesis as the mediator of signaling from thrombopoietin, erythropoietin and GM-CSF receptors. JAK2 activates several signaling pathways including STAT transcription factors, phosphoinositide-3 kinase (PI3K) and mitogen activated protein kinase (MAPK) pathways, which promote cell proliferation. In MPN, JAK2 signaling is constitutively activated by mutations in JAK2, MPL or CALR. The central role of JAK2 signaling in MPN has led to the development of JAK2 inhibitors which act as ATP mimetics and stabilize JAK2 in the active form (type I inhibitors). Despite certain benefits, type I JAK inhibitors fail to reduce the MPN clone suggesting limited disease-modifying potential and induce resistance. To improve therapeutic targeting of JAK2 signaling, we are pursuing several approaches:
Targeting MAPK pathway activation
We found that MAPK signaling remains activated in MPN despite JAK2 inhibition. This suggests that compensatory activation of MAPK effectors bypass pharmacologic JAK2 blockade. We identified PDGFR signaling as a mediator of compensatory MAPK activation and dual targeting of JAK2 and MEK1/2, intermediate kinases in the MAPK pathway, showed superior therapeutic efficacy. Correction of splenomegaly and cytoses was improved by combined JAK2/MEK inhibition. Bone marrow fibrosis was reduced to an extent not seen with JAK2 inhibitors suggesting the MAPK pathway needs to be targeted for enhanced therapeutic efficacy.
More effective targeting of JAK2
A new mode of JAK2 inhibition has been reported which stabilizes the inactive form of JAK2 (type II inhibitors). We found high potency of type II JAK inhibition in preclinical MPN models. We observed decreased allele burden suggesting type II JAK inhibition could lead to a class of agents with disease-modifying potential. Resistance to type I JAK2 inhibitors is also abrogated. We are continuing our studies on type II JAK2 inhibition given the potential to lead to more potent, clone-directed JAK2 inhibitors.
Mechanisms of resistance to JAK2 inhibitors
Response to type I JAK2 inhibitors may be lost upon prolonged exposure, but JAK2 resistance mutations have not been found in patients. It has been shown that MPN cells reactivate JAK2 signaling by formation of JAK2 heterodimers with other JAK family members as JAK1 and TYK2. We found that this escape mechanism extends to type I JAK inhibitors in clinical development and observed crossresistance. These molecular studies of resistance to JAK inhibitors may reveal new therapeutic targets, while studies on patient samples will provide insight into clinical JAK2 inhibitor resistance.
JAK2 signaling network
JAK2 activates STAT-, PI3K- and MAPK pathways and we have seen that combined inhibition of e. g. JAK2/Bcl2 or JAK2/MEK can increase benefit in JAK2-driven leukemia. We are investigating the JAK2 signaling network to delineate mechanisms limiting efficacy of JAK2 inhibitors to inform therapeutic strategies. We aim to extend these studies to other myeloid malignancies with suboptimal clinical benefit of tyrosine kinase inhibitors.
JAK2 signaling in thrombopoiesis
Efficacy of therapeutic targeting with JAK2 inhibitors is limited by on- and off-target toxicities. Thrombocytopenia is a relevant side effect of JAK2 inhibition. We have shown that JAK2 regulates megakaryopoiesis including formation of megakaryocyte-biased stem cells and are interested in differential effects of JAK2 inhibitors on thrombopoiesis.
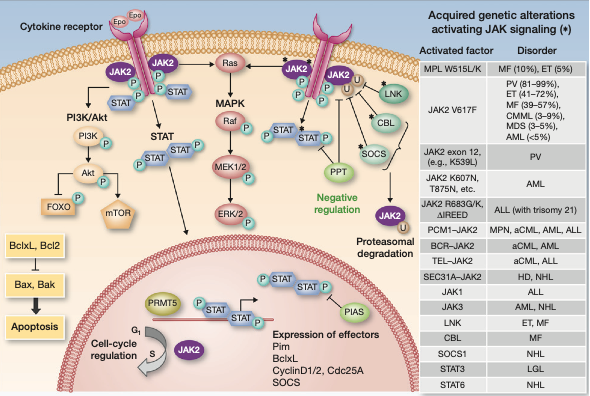
Fig. 1: Overview of JAK2 signaling. The JAK2 tyrosine kinase associates with hematopoietic cytokine receptors for EPO, TPO and GM-CSF. Upon ligand binding, JAK2 activates several signaling pathways including the STAT3 and STAT5 transcription factors, the PI3K/Akt pathway and the MAPK signaling pathway which includes RAS and the kinases RAF, MEK1/2 and ERK1/2. In MPN, JAK2 is constitutively activated by somatic mutations leading to excessive myeloid proliferation. The molecular interconnections between JAK2 and the downstream signaling pathways is not fully clarified. (Adapted from Meyer SC & Levine RL, Clin. Cancer Res. 2014)
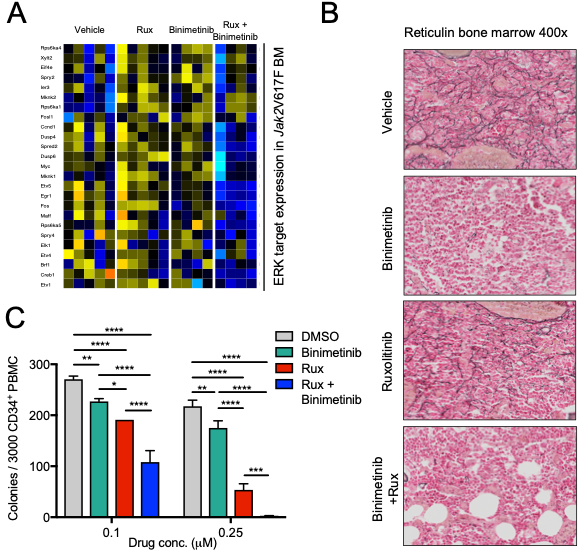
Fig. 2: Combined targeting of JAK2 and MEK provides therapeutic benefit in MPN in vivo. Pharmacologic inhibition of MEK by binimetinib and JAK2 by ruxolitinib is superior to ruxolitinib alone in reducing ERK target expression (A) and fibrosis in Jak2V617F mouse bone marrow (B). Combined JAK2/MEK inhibition is also superior to ruxolitinib monotherapy in suppression of myeloid colony formation from MPN patient CD34+ cells (C, adapted from Stivala S, Codilupi T, Brkic S, Meyer SC. J Clin Invest 2019.).