3D Printing . OPA1 . Autosomal Dominant Optic Atrophy . Artificial Transcription Factor Fuchs’ Dystrophy . Corneal Implant
Ocular Pharmacologyand Physiology
Translational research to treat blinding diseases
Visual impairment and the associated socio-economic impact affects an estimated 290 million people worldwide. Our research is aimed at understanding the causes of vision loss and to develop novel therapies to prevent or treat blinding diseases.
Treating autosomal dominant optic atrophy
The optic nerve formed by the axons of retinal ganglion cells (RGCs) relays visual information form the light-sensing retina to the vision centers in the brain. Thus, loss of RGCs results in visual impairment and ultimately blindness.
Autosomal dominant optic atrophy (ADOA) is the most common inherited form of optic nerve degeneration. Mainly caused by mutations in the OPA1 gene, visual impairment develops in the first decade of life and can progress to complete blindness. The OPA1 gene codes for a mitochondrial protein involved in maintaining mitochondrial morphology by modulating mitochondrial fusion. In addition, OPA1 is essential for mitochondrial cristae organization and, thus, electron transport chain function. Loss of OPA1 is known to diminish mitochondrial fidelity and to increase sensitivity to apoptotic stimuli. Critically, OPA1 mRNA is subject to extensive alternative splicing giving rise to at least eight different OPA1 isoforms with a balanced isoform expression important for OPA1 function.
About 70 % of ADOA cases are caused by OPA1 haploinsufficiency. Using artificial transcription factors (ATFs) capable of upregulating OPA1 expression, we aim to alleviate OPA1 haploinsufficiency while maintaining OPA1 isoform balance, thus providing a functional cure for ADOA. Based on TAL effectors, we generated ATFs capable of upregulating mouse or human OPA1 (Fig. 1). To test efficacy of such ATFs, we generated a novel in vivo model of pharmacologically accelerated RGC degeneration in ADOA mice. This allowed us to synchronize RGC death and shortened the time it takes to detect treatment efficacy from 12-15 months to 14 days. Currently, we are in the process of optimizing viral delivery to allow for efficient delivery of OPA1-upregulating ATFs to RGCs by simple intravitreal injection.
Towards a treatment for Fuchs’ dystrophy
Vision starts with light entering the eye through a transparent cornea. The cornea is a five-layered tissue with the outermost corneal epithelium together with Bowman’s layer and the innermost corneal endothelium with Descemet’s membrane sandwiching the almost cell-free corneal stroma. To keep the corneal stroma transparent, corneal endothelial cells (CECs) actively remove water from this tissue. During Fuchs’ dystrophy and due to CEC loss, water accumulates inside the stroma causing a disorganization of the carefully arranged stromal matrix leading to severe visual impairment. Interestingly, about 4 % of the population over 40 years of age suffers from Fuchs’ dystrophy to a varying degree. Severe Fuchs’ dystrophy is treated by replacing the damaged CEC layer with human donor material in a procedure called Descemet Membrane Endothelial Keratoplasty (DMEK). However, the demand for corneal transplants outweighs availability about 70:1.
By combining additive manufacturing and stem-cell technology, we want to address this unmet medical need by generating a drop-in replacement for human donor material for DMEK. We employ melt electrowriting to fabricate a suitable scaffold for culturing human CECs (Fig. 2). Furthermore, a differentiation protocol for CECs from induced pluripotent stem cells (iPSCs) was developed and adapted to be compatible for culturing these cells on such scaffolds. To assess whether these implants are compatible with the established transplantation procedure, cell-free scaffolds were implanted into porcine eyes ex vivo. Imaging revealed that the fabricated structural support is well suited as drop-in replacement during DMEK. Taken together, with developing a donor tissue replacement for DMEK we hope to address the unmet medical need of patients suffering from Fuchs’ dystrophy.
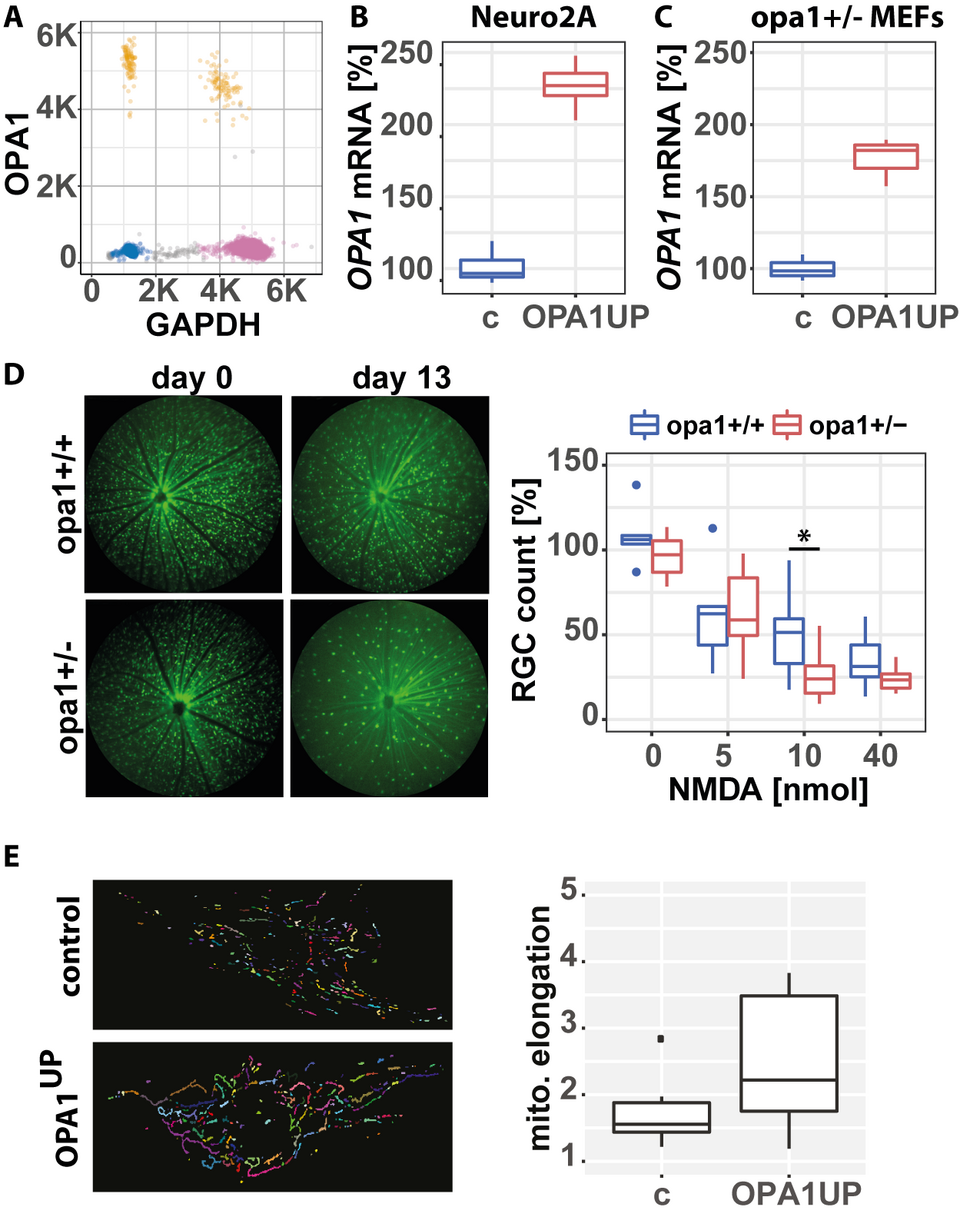
Fig. 1: Xanthomonas transcriptional activator like (TAL)-based artificial transcription factors (OPA1UP) capable of upregulating OPA1 in mouse or human cells was generated. (A) To accurately measure OPA1 mRNA levels, a digital droplet PCR (ddPCR) assay was established. Following expression of OPA1UP fused to T2A-GFP, GPF+ cells were sorted and multiplexed measurement of OPA1 and GAPDH mRNA levels was possible in as little as 100 cells. (B) Mouse neuron-like Neuro2A and (C) opa1+/- mouse embryonic fibroblasts (MEFs) were transfected with expression plasmid for OPA1UP-T2A-GFP, GFP+ cells were isolated by fluorescence activated cell sorting and OPA1 mRNA levels were quantified relative to GAPDH. Please note, opa1+/- MEFs are haploinsufficient for OPA1. (D) OPA1+/- mice with GFP-expressing retinal ganglion cells (RGCs) and their wildtype littermates were treated with increasing amounts of NMDA by intravitreal injection. Fluorescent fundoscopy was performed before and 13 days after NMDA application. Please note the significantly lower amount of surviving RGCs in opa1+/- animal compared to wildtype controls following application of 10 nmol of NMDA (n>15 animal/condition). (E) Patient-derived, primary human fibroblasts transfected with OPA1UP or inactive control were stained for the mitochondrial marker cytochrome c and mitochondrial morphology was analyzed. Please note the increase in mitochondrial length following OPA1UP expression compared to controls indicative for restored OPA1 function.
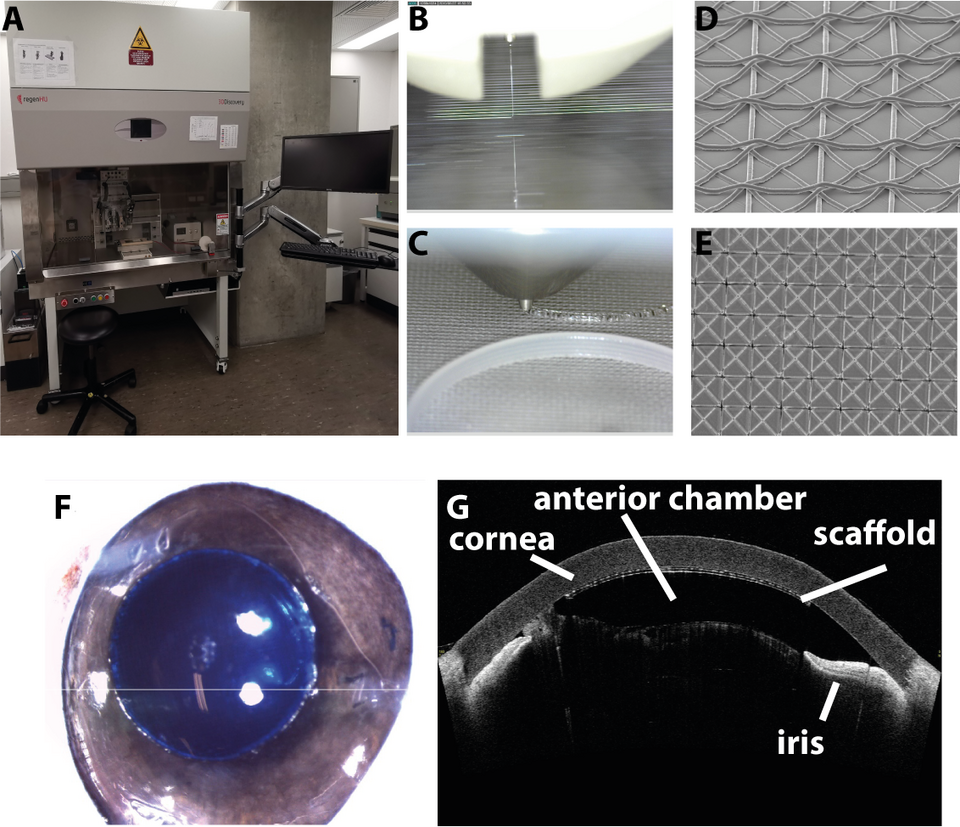
Fig. 2: (A) Precision 3D printer (regen-HU, 3D Discovery) equipped with four printheads including a melt electrowriting head capable of depositing molten polymer with a lower diameter of 600 nm in a highly reproducible fashion. (B) Microscopic image of molten poly-caprolactone (PCL) deposited onto a glass collector plate in a 6000 KV electric field to form 10 μm fibers. (C) Deposition of PCL support structures onto melt electrowritten scaffold using a fused deposition printhead. (D+E) Electron microscopic images of melt electrowritten PCL fibers. (F) A PCL scaffold embedded into artificial extra-cellular matrix (stained blue for better visualization) was implanted ex vivo into a porcine eye using the standard surgical procedure to perform Descemet Membrane Endothelial Keratoplasty (DMEK). (G) Anterior chamber optical coherence tomography was used to assess placement and attachment of the implanted scaffold.