Innate Immunity . Inflammation . Arthritis, Macrophage . Micro RNA. Mitochondrial DNA
Experimental Rheumatology
The role of innate immune mechanisms in the pathogenesis of chronic inflammatory rheumatic diseases
The research of the Experimental Rheumatology (ER) group is focused on the pathogenetic mechanisms of inflammatory rheumatic diseases including chronic arthropathies such as rheumatoid arthritis (RA) and psoriatic arthritis (PsA), connective tissue diseases such as systemic lupus erythematosus (SLE), Sjögren’s syndrome (SS) and systemic sclerosis (SSc), autoimmune vasculitides (AAV) as well as the crystal arthropathies gout and calcium pyrophosphate deposition disease. In several research projects, including collaborations with academia and pharma/biotech industry, we investigate the immunopathogenesis of some of these diseases with special focus on mechanisms of the innate immunity.
The research topics include
– the regulation of inflammatory pathways during macrophage differentiation and polarization.
– immune-modulation by extracellular vesicles and the identification of their functional load in rheumatoid arthritis.
– the screening of novel inhibitors targeted against inflammatory pathways and autoimmune disease- relevant factors
– the role of neutrophil extracellular traps and cell-free mitochondrial DNA in the pathogenesis of various rheumatic diseases.
We follow a translational approach, using patient materials such as blood and synovial fluid. In vitro cell cultures with primary cells are set up and phenotypic and functional analyses are performed in vitro. In our recent work one of the main topics was the analysis of microRNA (miR) as epigenetic regulators of inflammatory signaling pathways (Figure 1). In a search for microRNA expressed by in vitro differentiated macrophages stimulated via TLR ligands we have identified miR-221-3p as a driver of a polarization of macrophages towards a proinflammatory M1 phenotype. We found that overexpression of miR-221-3p in anti-inflammatory M2-macrophages is repressing JAK3/STAT3 signaling that governs anti-inflammatory IL-10 secretion in these cells. In addition, miR-221-3p overexpression or pharmacological inhibition of JAK3 not only suppressed IL-10 secretion but also imposed a pro-inflammatory cytokine profile in M2- macrophages, including an increased secretion of IL-12 and IL-6. We hypothesize that this mechanism may contribute to chronic inflammation and destruction in rheumatoid arthritis (RA). Our results add to existing evidence suggesting microRNAs as tarpeutic targets. Currently we are involved in an Innosuisse funded collaborative project searching for small molecular inhibitors of microRNA. Another focus is the role of neutrophil extracellular traps and cell-free mitochondrial DNA in the pathogenesis of various rheumatic diseases. Activated neutrophils have been implicated in the pathogenesis of Systemic Lupus Erythematosus (SLE) and the ANCA-associated vasculitides (AAV). Recent data suggests that subjects with SLE are characterized by impaired NET degradation, disseminating the availability of extracellular DNA as a pro-inflammatory stimulus to the innate immune system. NETs may also contain mitochondrial DNA (mtDNA), a dsDNA molecule which is phylogenetically evolved from bacteria and rich in hypomethylated CpG sequences, thus especially suited to trigger TLR9 signaling and disease flares. The main goal of the particular study is to analyse the extent and nature of circulating extracellular DNA species (mtDNA vs. nDNA) in SLE and AAV and determine if mtDNA plasma concentrations can serve as a marker for diagnosis and disease activity. Overall we aspire to contribute to a better understanding of the contribution of innate immune mechanisms to the pathogenesis of rheumatic diseases.
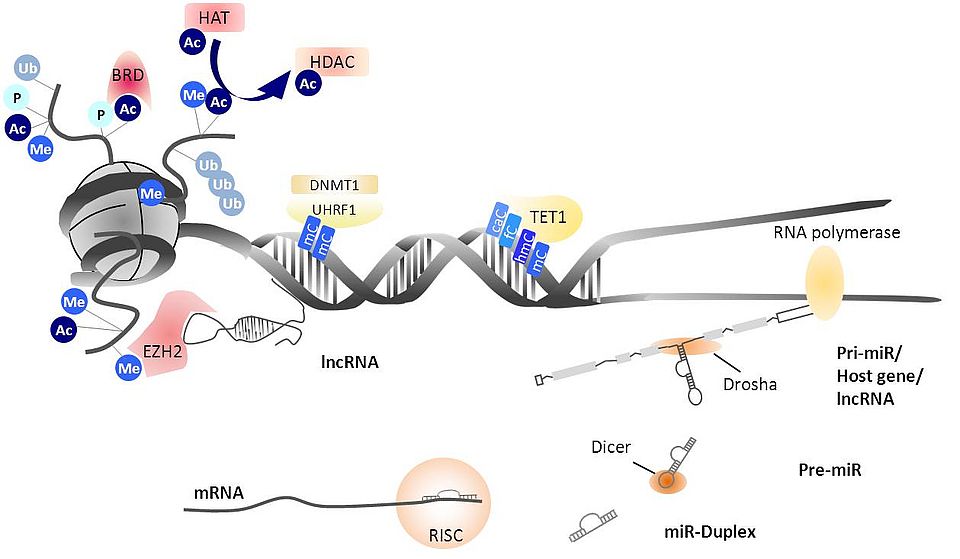
Fig. 1: Epigenetic modifications and microRNA biogenesis. MicroRNAs are transcribed as primary transcripts (pri-miR) that can be embedded in host genes or lncRNA. Drosha processes the pri-miR and Dicer cleaves the resulting precursor miR (pre-miR). The miR-Duplex is loaded onto the RNA induced silencing complex (RISC) where the separated strands bind their target mRNA. Ac = acetylation, Me = methylation, P = phosphorylation, Ub = ubiquitination Modified from: Kyburz, Karouzakis and Ospelt, Best Pract Res Clin Rheumatol (28) 2014: 577–587