Genome Plasticity . DNA Damage and Repair . DNA Methylation and Demethylation Epigenetic Control of Genome Function
Genome Plasticity
Genome dynamics in cell programming, aging and human disease
DNA in our cells is continuously damaged through its exposure to reactive agents of endogenous or environmental origin. Damage to DNA, however, does not only occur randomly by chemical reactions, but also on purpose, by the action of enzymes, to increase genetic variance or alter cell fate determining epigenetic marks, i.e., DNA methylation. Modifications of either kind occur thousands of times in our DNA every day and need to be controlled if genome integrity is to be maintained. We investigate molecular mechanisms underlying this dynamic instability of genomes. Over the past years, our research focus has been the role of DNA repair in DNA demethylation and its contribution to the patterning and maintenance of epigenetic programs – hence – cell identity. We have been following three main lines of investigation towards (i) unraveling the basic molecular mechanisms and function of active DNA demethylation, (ii) the relevance of DNA methylation control and stability for human aging and disease, and (iii) the impact of the environment on the stability of DNA methylation.
Shaping chromatin by DNA repair mediated DNA demethylation
An important longterm interest of our research has been a DNA repair pathway controlled by the “Thymine DNA Glycosylase” (TDG). TDG first caught our attention because of its ability to remove thymine (T) or uracil (U) when mismatched with guanine (G) in DNA. T•G and U•G mismatches arise frequently by hydrolytic deamination of cytosine (C) or 5-methylC (5mC) and, unless repaired, will give rise to C>T mutations, the most prevalent base substitution found in our genomes. While its enzymatic activity implicates TDG in the anti-mutagenic repair of these mismatches, our research led us to uncover a quite different function. It started with the unexpected observation that a genetic defect in TDG causes developmental failure in a mouse model, due to aberrant gene expression and DNA methylation patterning. Together with work of others on the biochemistry of transeleven-translocation (TET) proteins, these findings indicated TDG to act in a pathway for active DNA demethylation, operating through the oxidation of 5mC by TET and replacement of the oxidized 5mC with a C through TDG dependent DNA repair. We then found that TDG and TET indeed cooperate in differentiating cells to drive cyclic methylation and demethylation at specific gene regulatory sequences. We were able to show that TET1 and TDG physically and functionally interact to form an active DNA demethylase, and to proof by full biochemical reconstitution that the TET-TDG-repair system, coordinated by SUMO modification, is capable of productive DNA demethylation. Ongoing work addresses, amongst other questions, the “biology” of TDG driven DNA methylation/demethylation cycling, in particular its implicated role in shaping chromatin and gene expression states in differentiating cells.
(Epi)genetic maintenance by DNA repair
Aberrant DNA methylation contributes to tumorigenesis by deregulation of the genome, but exactly why and how such aberrations arise is largely unknown. Our aim is to identify and mechanistically understand genetic und environmental conditions modulating DNA methylation stability in human tissues. Investigating DNA methylation stability in the human colonic mucosa with a molecular epidemiological approach, we were able to decipher distinct patterns of age-dependent DNA methylation drift and found the rate of such DNA methylation change to be modulated by lifestyle factors, including BMI, medication and smoking. This work in healthy tissue allowed us then to derive cancer-specific patterns of aberrant DNA methylation and, subsequently, to separate on that basis subtypes of colon cancer that evolve through different molecular pathways associated with and without a CpG-island methylator phenotpype (CIMP). Mechanistically, we were able to show CIMP in colon cancer to be a consequence of a malfunction in TET dependent active DNA demethylation, caused by BRAF-induced downregulation of TET1. Current work addresses the mechanism underlying this link between oncogenic signalling an epigenetic remodelling.
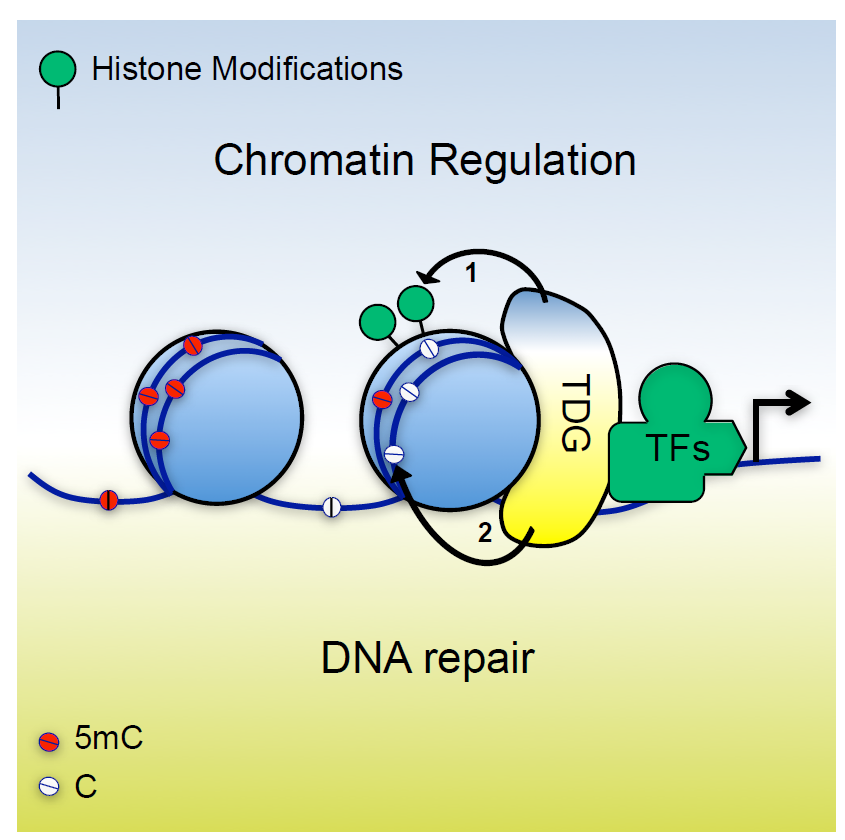
Fig. 1: TDG dependent DNA excision repair controls epigenetic states through DNA demethylation.
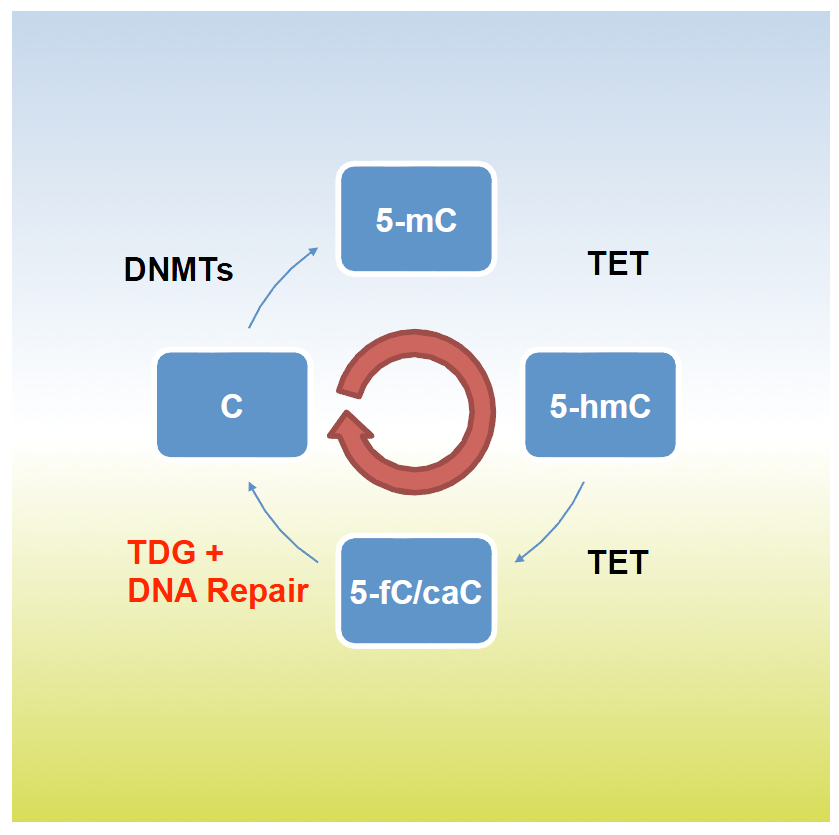
Fig. 2: TDG and TET hydroxylases cooperate in cyclic DNA methylation and active oxidative demethylation at CpG di-nucleotides in the genome. TDG excises 5-fC and 5-caC, thereby initiating excision repair incorporating an unmethylated C. 5-mC, 5- methylcytosine; 5-hmC, 5-hydroxymC; 5-fC, 5-formylC; 5-caC, 5 carbocylC.
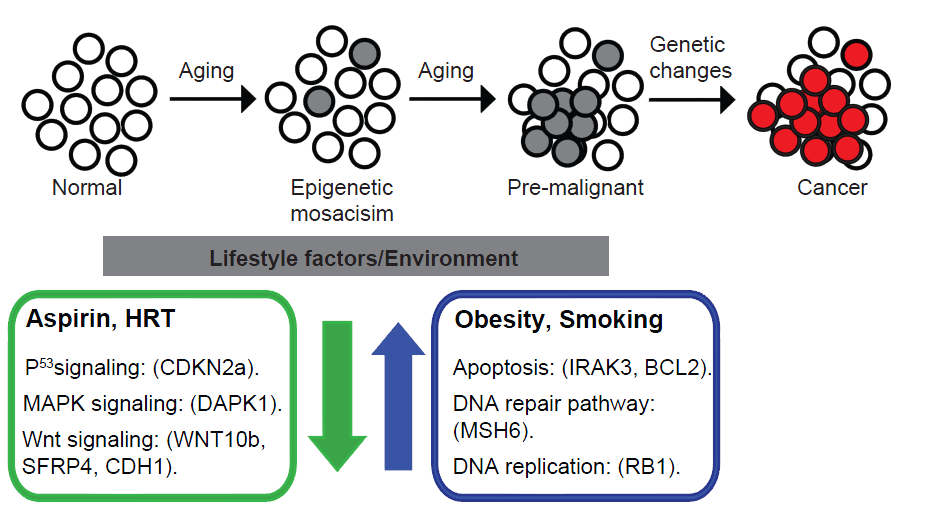
Fig. 3: Lifestyle factors modulate the rate of DNA methylation drift in the aging colonic mucosa and, by inference, early events of colorectal carcinogenesis.