Acute Myeloid Leukemia . Models . Mechanisms . Targets . Therapeutic strategies
Childhood Leukemia
Models and molecular mechanisms of acute myeloid leukemia (AML)
Our research aims to understand the cellular and molecular mechanisms of a human blood cancer called acute myeloid leukemia (AML). We focus on genetic lesions associated with particularly aggressive disease that, despite improved therapeutic options, remains incurable for most of the patients. Based on the rarity and genetic heterogeneity of the disease with limited access to primary material, we perform most of our studies in cell- and mouse models. During the last three years, we worked on two major subjects.
A) We studied the impact of the cellular origin and transforming potential of AML-associated fusion oncogenes by generating a series of inducible transgenic mouse lines (in collaboration with A. Peters, FMI, Basel and T. Mercher, Institute Gustave Roussy, Paris). We found that these fusions act as epigenetic regulators and induce a reversible leukemia in mice that closely phenocopies the human disease. Interestingly, induction of an MLL-AF9 fusion in hematopoietic stem cells (HSC) resulted in a more aggressive disease than activation in more committed progenitor cells. In contrast to MLL-AF9, the MLL- ENL fusion only resulted in leukemia when induced in HSC and early multipotent progenitors suggesting a particular window of hematopoietic transformation susceptibility (Stavropoulou et al., 2018). The NUP98-MLL fusion is the only AML-associated MLL rearrangement in which the entire C-terminus of the protein is maintained. Induction of NUP98-MLL resulted in myelodysplastic syndromes and AML in mice (Fisher et al., 2020). The ETO2-GLIS2 fusion gene is a molecular hallmark of acute megakaryoblastic leukemia (AMKL) almost exclusively affecting young children. Strikingly, ETO2-GLIS2 induction in mouse fetal HSC rapidly induced reversible AMKL, while expression in adult mouse BM cells resulted in long latency AML. Chromatin and transcriptomic analysis revealed that ETO2-GLIS2 rewired transcription factor activities in an ontogeny-dependent manner. Thus, we established the first in vivo model for ETO2-GLIS2-driven AMKL and showed that the phenotype of pediatric AML is determined by ontogeny-dependent susceptibility for transformation by particular oncogenic fusion genes (Lopez et al., 2019).
B) Functional characterization of the nuclear interacting SET domain protein 1 (NSD1), a histone methyltransferase involved in a fusion oncogene in pediatric AML, led us to study acute erythroleukemia (AEL). Genetic inactivation of NSD1 in the hematopoietic system of the mouse unexpectedly resulted in development of a fully penetrant disease with several hallmarks of human AEL. Functional studies revealed that despite abundant expression of the erythroid transcriptional master regulator called GATA1, differentiation of Nsd1-/- erythroblasts was significantly impaired. Expression of wildtype but not a catalytically-inactive NSD1 mutant rescued differentiation associated with increased GATA1 chromatin occupancy and target gene activation. Collectively our work indicates that the NSD1 methyltransferase is a novel regulator of GATA1-controlled erythroid differentiation and leukemogenesis (Leonards et al., 2020). AEL is a rare but very aggressive human cancer. To better understand its molecular pathology, we genetically characterized a series of world-wide collected primary AEL samples. We found at least three molecular subgroups including patients carrying TP53 mutations, epigenetic mutations, and others. We established a transcriptomics-based space in which, independently of the genetic subgroup, most AEL samples exhibited a unique mapping that was clearly different from other AML or MDS. In >25% of the cases, we found aberrant expression of transcriptional regulators related to impaired activity of GATA1. Expression of these factors immortalized murine erythroid progenitors in vitro and led to erythroid or erythroid/myeloid proliferations in vivo, phenocopying human AEL. This work indicates that AEL is a genetically heterogeneous disease with an erythroid identity that results in part from the aberrant activity of key erythroid transcription factors like GATA1 (Fagnan et al., 2020).
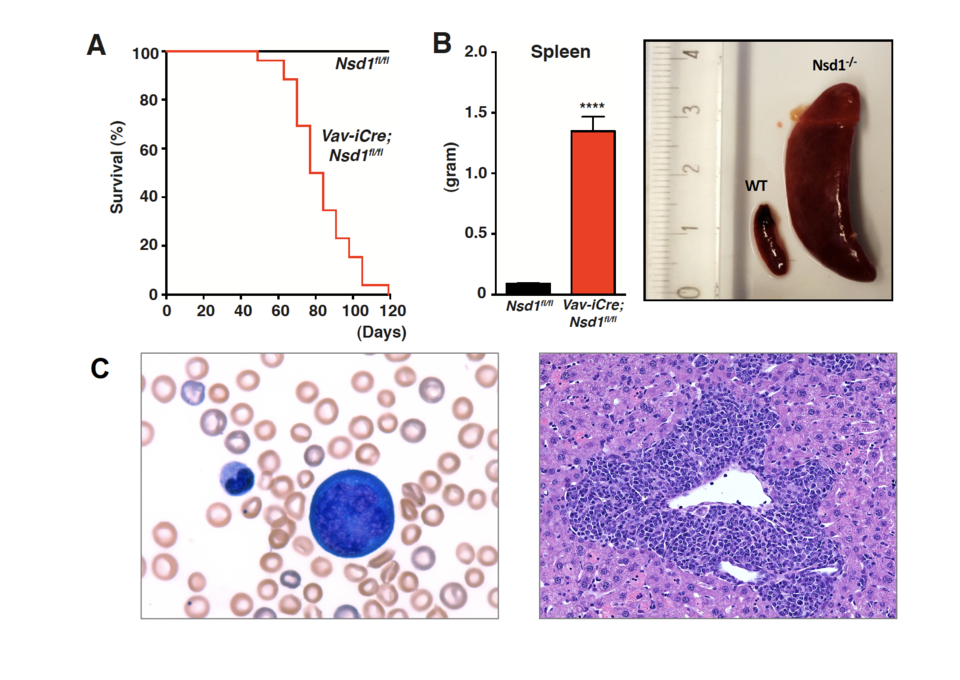
Fig. 1: Genetic inactivation of the NSD1 histone methyltransferase in the hematopoietic system of the mouse results in a fully penetrant disease that phenocopies human acute erythroleukemia. A) Kaplan-Meier survival plot B) Significant splenomegaly of diseased Vav-iCre;Nsd1fl/fl (= Nsd1-/-) mice C) Left: peripheral blood smear of a diseased Nsd1-/- mouse with a large dysplastic multinuclear erythroblast, a smaller myelocyte, reticulocytes and poorly globinized erythrocytes; right: liver section showing extensive infiltration by erythroblasts.