Neuromuscular Research
Our neuromuscular research laboratory is part of the Clinic for Neurology and the Department of Biomedicine and focuses on the elucidation of pathophysiological mechanisms of neuromuscular diseases and on the development of new therapies for patients affected by muscle disorders.
In particular, our research interests focus on the development of therapeutic strategies for the common muscular dystrophies myotonic dystrophy, facio-scapulo-humeral muscular dystrophy and congenital myopathies. Our research also aims to elucidate the pathophysiological mechanisms underlying congenital myopathies and the pharmacogenetic disorder malignant hyperthermia, caused by mutations in RYR1 the gene encoding the ryanodine receptor 1. More specifically we are interested in deciphering how mutations in RYR1 affect calcium homeostasis and cause diseases. We investigate how individual gene profiles influence the effect of anesthetics and how we can improve diagnosis and patient safety. We are also interested in the pathophysiological mechanisms that lead to muscle breakdown in neuromuscular diseases.
Department of Biomedicine
ZLF LAB 408
University Hospital Basel
Hebelstrasse 20
4031 Basel
Myotonic Dystrophy . RNA Splicing . FSHD . DUX4. Muscle Wasting . Proteostasis . E3 ubiquitine ligase
Translational Research in Neuromuscular Diseases (Sinnreich)
Our Neuromuscular Research Laboratory is part of the Clinic of Neurology and the Department of Biomedicine and focuses on the elucidation of pathophysiological mechanisms involved in neuromuscular diseases and on the development of therapeutic strategies.
Myotonic dystrophy type I (DM1) is a disabling neuromuscular disease with no causal treatment available. It is the most prevalent muscular dystrophy in adults, affecting about 1 in 10’000 individuals. This disease is caused by expanded CTG trinucleotide repeats in the 3’ UTR of the dystrophia myotonica-protein kinase gene (DMPK). On the RNA level, expanded (CUG)n repeats form hairpin structures that sequester splicing-factors, such as muscleblind-like 1 (MBNL1). Lack of available MBNL1 leads to mis-regulated alternative splicing of many target pre-mRNAs, causing multisystemic involvement in DM1. In an effort to identify small molecules that liberate sequestered MBNL1 from (CUG)n RNA, we developed a pathomechanism-based screening cascade including biochemical, cellular and animal model assays which allow for high throughput screening of small molecular weight compounds. Identified hits may provide pharmacophores for further medicinal chemistry optimization.
Facio-scapulo-humeral muscular dystrophy (FSHD) is the second most common muscular dystrophy in adults, affecting about 1:20’000 persons. An epigenetic aberration leads to the ectopic expression of the transcription factor Double Homeobox protein 4 (DUX4) in skeletal muscle and other tissues, which leads to muscle cell degeneration and muscular dystrophy, sensorineural hearing loss and retinal teleangiectasias. Expression of DUX4 variants are also involved in certain cancers including acute lymphoblastic leukemia. An attractive therapeutic approach would be the interference with aberrantly expressed DUX4.
By applying Systematic Evolution of Ligands by Exponential Enrichment (SELEX) and fluorescence-based biochemical assays we were able to generate a DNA aptamer with high affinity towards DUX4. In a collaborative effort we co-crystallized DUX4 together with the identified oligonucleotide enabling us to explain the affinity boost caused by certain bulge loops. We plan to use this oligonucleotide as a tool to further study DUX4-DNA interactions and to develop treatment strategies for FSHD and other DUX4-mediated diseases.
An additional line of research in our laboratory is dedicated to the identification of mechanisms underlying disruption of proteostasis as a cause for muscle diseases. To this end, we initiated experimental strategies to identify molecular networks that specifically control protein synthesis and degradation in human myofibers under physiological conditions and upon atrophic stress. Our strategy integrates the genome-wide measurements of a.) transcript levels (RNA-Seq), b.) translation levels (sequencing of ribosome foot prints) and c.) protein levels (proteomics) from genetically (Crispr/Cas9) modified human immortalized myoblasts. The ribosome foot-printing method, which we developed together with the laboratory of Prof. M. Zavolan, provides us with estimates of protein synthesis rates of individual RNA molecules, thus allowing us to further uncover the regulatory factors of translation for each gene under normal and diseased conditions. In parallel, we developed in situ proximity-dependent labelling assays (BioID) to map the direct and indirect interactome of the two main proteins we have already identified in skeletal muscle atrophy, MAFbx and EIF3F (Fig. 3).
Connection to Clinical Practice
Interdisciplinary Neuromuscular Clinic
At our interdisciplinary Neuromuscular Clinic we care for patients affected by a broad range of neuromuscular diseases. In collaboration with our colleagues from pathology, genetics, plastic surgery, pulmonary medicine, rehabilitation, ergo-, physio- and speech therapy as well as social services, we provide clinical and electrophysiological evaluation, perform muscle and nerve biopsies with histopathological and biochemical workup, genetic workup and counseling, rehabilitation, ergo-/physio- and speech therapy as well as assistance in social matters. Novel clinical observations are being worked up scientifically and form the basis for translational research projects.
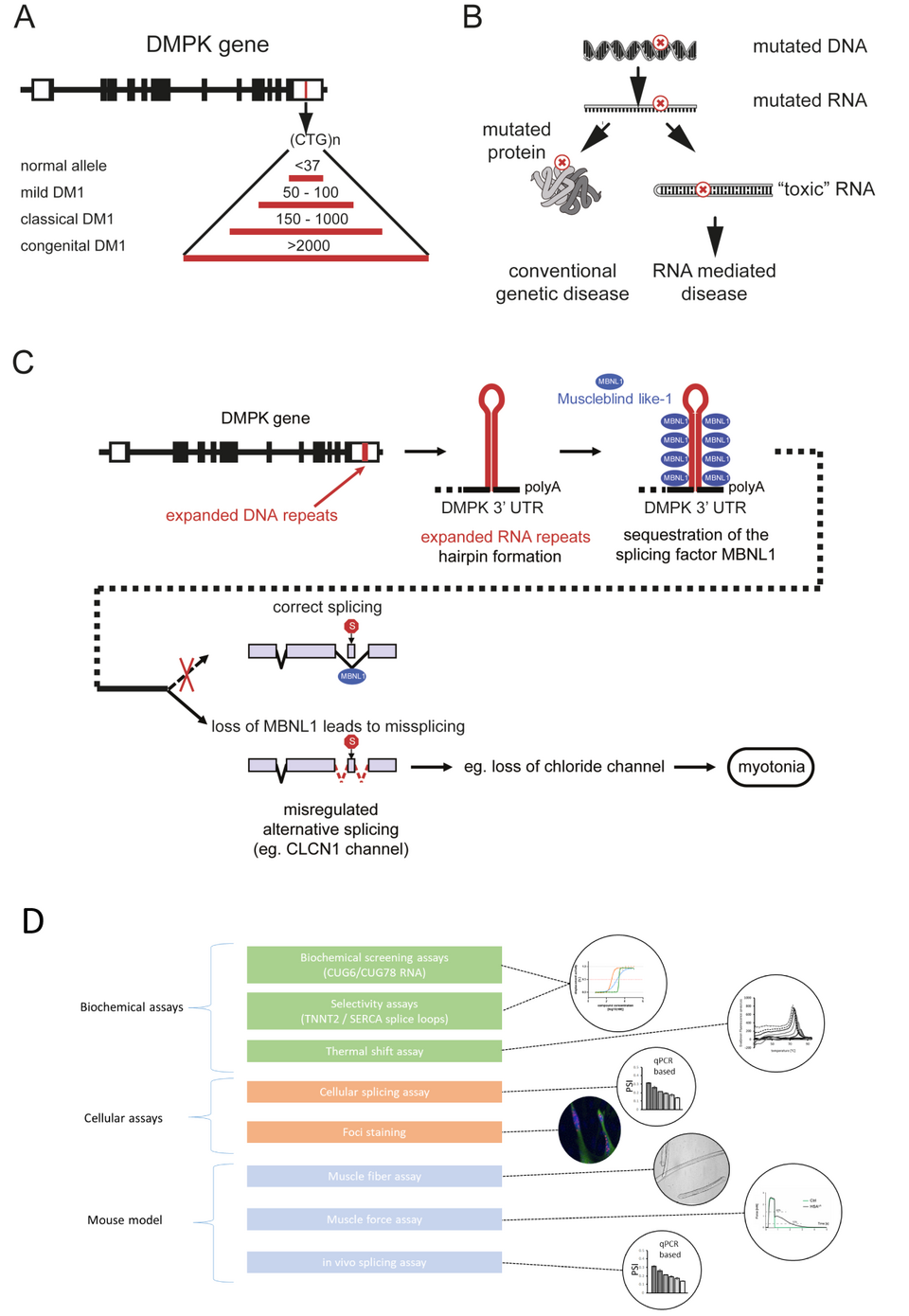
Fig. 1: Pathophysiology of DM1 and screening cascade to identify small molecular weight compounds. A) The molecular basis of DM1 is an expansion of an unstable repeat sequence in the noncoding part of the DMPK gene. Severity of disease is correlated with the size of the repeat expansion. B) In DM1, the mutation is located in a noncoding region and does not alter the protein sequence, but leads to toxic RNA. C) The sequestration of the alternative splicing factor MBNL1 by toxic RNA leads to altered splicing of target pre-mRNAs like CLCN1, encoding muscle-specific chloride channel (ClC-1). This mis-splicing leads to ClC-1 deficiency and to myotonia. D) We established a pathophysiology based screening cascade including biochemical, cellular and animal model assays to identify small molecular weight compounds able to disrupt the interaction between MBNL1 and the toxic RNA, and to restore splicing and function.

Fig. 2: Generation of a high affinity aptamer against DUX4. A) Predicted secondary structure of the high affinity oligonucleotide. DUX4 binding motif is highlighted in red (forward motif) and blue (reverse motif). Affinity bulge loop is highlighted in yellow. B) KD value for DUX4–oligonucleotide binding, determined by fluorescence polarization assay. Data are shown with 95 % confidence band in gray. Fitting results are displayed in the table at the upper left corner. C) Crystal structure of DUX4 double homeodomain bound to the DNA oligonucleotide containing a trinucleotide (-CCC-) bulge and a GCA hairpin loop. DNA is shown in magenta. DUX4 is colored in a gradient of blue to red from the N- to C-terminus, respectively. D) A close-up view showing the DUX4 interactions with the CCC bulge loop. The guanidinium group of Arg79 stacks onto the first C base of the bulge (the van der Waals or cation-π-contact is indicated by a red dotted line). Arg80 forms a salt bridge with the third C base and Arg76 is hydrogen-bonded to a DNA backbone phosphate at the 3’-end of the bulge (yellow dotted lines)
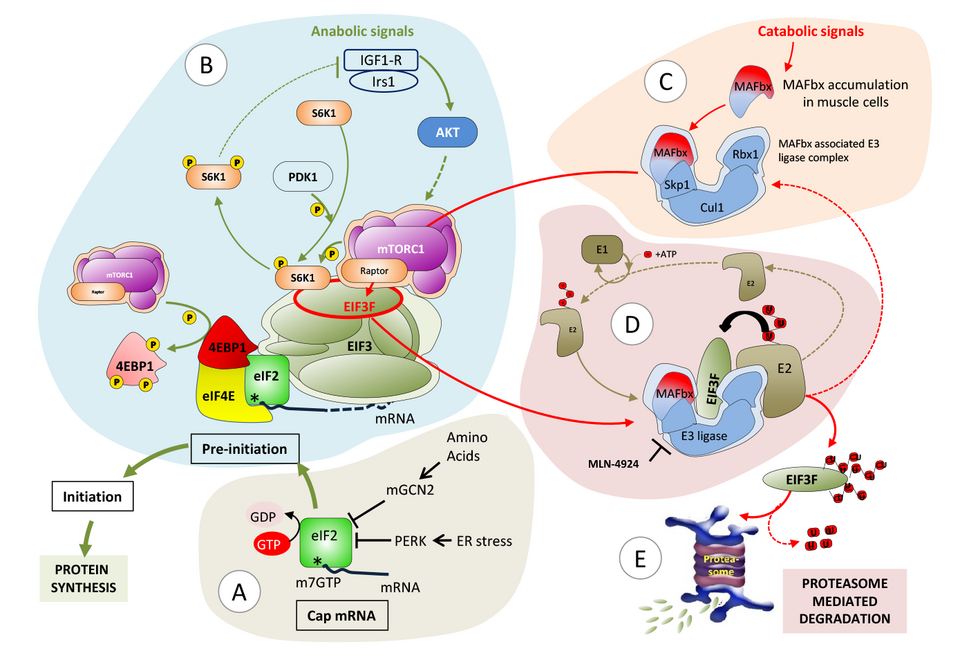
Fig. 3: Proposed model for modulation of protein synthesis in skeletal muscle. A) Assembly of capped mRNA and integration of stress signaling. B) EIF3F dual function: protein initiation complex (PIC) recruitment at the mRNA cap and scaffolding platform for mTORC1 and S6K1. mTORC1 affects protein synthesis via phosphorylation of 4EBP1, which thereby dissociates from eIF4E; and phosphorylation of S6K1, which facilitates the release of S6K1 from EIF3F and its activation by PDK1. C) Atrophy dependent induction of MAFbx leads to the formation of MAFbx E3ligase complex together with Cul1, Skp1 and Rbx1. D) MAFbx dependent EIF3F ubiquitination followed by E) proteasome mediated degradation.
Ryanodine Receptor . Mutations . Calcium . Skeletal Muscle . Congenital Myopathies Mouse Models
Functional effects of ryanodine receptor mutations linked to congenital muscle diseases (Treves)
In skeletal muscle calcium is a key second messenger regulating contraction and the sarcoplasmic reticulum (SR) is the intracellular organelle involved in its regulation. The ryanodine receptor Ca2+ channel (RyR1) present on the terminal cisternae of the SR, is closely apposed to and is directly activated by, the dihydropyridine receptor an L-type Ca2+ channel functioning as voltage sensor. Upon depolarization, the voltage sensor undergoes a conformational change promoting the opening of the RyR1, leading to Ca2+ release from the SR; this elevation of the myoplasmic [Ca2+] is necessary for, and leads to, muscle contraction and this process is called excitation-contraction coupling (Fig. 1).
More than 700 RYR1 variants have been identified in patients worldwide, making this gene the primary target of neuromuscular disorders and accounting for over 30 % of mutations found in patients with congenital myopathies. Both dominant and recessive RYR1 mutations occur and usually associate with different phenotypes. Dominant mutations are causative of malignant hyperthermia/rhabdomyolysis/ exertional heat intolerance and central core disease and functionally impact the channel’s biophysical properties. Patients bearing recessive mutations are generally more severely affected, characteristically also display involvement of extraocular muscles and are diagnosed as having multi-minicore disease/centronuclear myopathy. The latter mutations have no major effect on the channel properties, but their presence is accompanied by profound biochemical changes in patients’ muscles, including a significant reduction of the RyR1 protein content and high levels of expression of class II histone de-acetylases. The reduced RyR1 levels in the SR membrane cause a decrease of calcium release during excitation contraction coupling, leading to weak muscles.
Our laboratory focuses on determining the functional effect of RYR1 mutations with the long-term goal of developing a pharmacological strategy to improve muscle function in patients. To do so we use two main experimental models: patientderived biological material and transgenic mouse models knocked in for mutations identified in patients. Muscle biopsies are evaluated biochemically and physiologically. Our results have demonstrated that muscle biopsies from patients carrying recessive RYR1 mutations show abnormally low RyR1 protein content, alteration of gene methylation and an increase in the content of chromatin modifying enzymes including class II histone de-acetylases and DNA methyl transferases. A mouse model we knocked in for a mis-sense mutation in one allele and a frameshift mutation in the other allele (DKI mouse, Fig. 2) exhibits severe muscle impairment, reduced levels of calcium release and disorganization of the muscle ultrastructure. Additionally, extraocular muscles from the transgenic mice have impaired excitation contraction coupling, as well as almost no EO-MyHC, the eye-muscle specific myosin heavy chain isoform. These results are consistent with the weak eye muscles of patients carrying recessive RYR1 mutations.
We are also interested in other aspects of skeletal muscle and in particular in identifying and validating the function of newly identified SR proteins. We have characterized a number of novel proteins including JP-45, junctate, SRP-27 and SRP-35; the latter is a membrane bound retinol-dehydrogenase converting retinol (Vitamin A) to all trans-retinaldehyde. Our results show that SRP-35 is involved in glucose metabolism, facilitating glucose uptake into skeletal muscle. We are currently investigating directly the role of Vitamin A in skeletal muscle physiology. Taken together the results of our studies will have important implications especially since they will promote the development of pharmacological therapies to improve the quality of life of patients with disorders leading to a decreased levels of RyR1 and may shed new information regarding the link between metabolic disorders and skeletal muscle glucose metabolism.
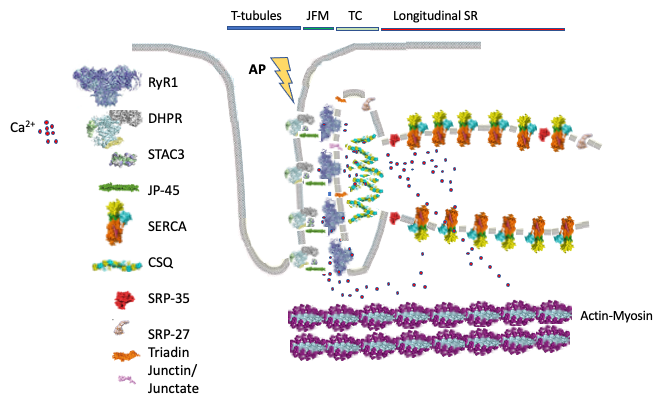
Fig. 1: Schematic representation of skeletal muscle proteins involved in excitation-contraction coupling. The figure shows the main components and subcellular localization of the proteins involved in skeletal muscle excitation–contraction coupling (ECC). The transverse (T-) tubules containing are invaginations of the plasma membrane where the voltage sensing dihydropyridine receptor (DHPR) is located. STCA3 binds to the DHPR. The T- tubules face the sarcoplasmic reticulum junctional0 face membrane (JFM) containing the ryanodine receptor 1 (RyR1) Ca2+ release channel, as well as JP-45, triadin and junctin/junctate/asparty β-hydroxylase. Calsequestrin bind Ca2+ and forms a mesh within the lumen of the sarcoplasmic reticulum. Opening of the RyR1 leads to Ca2+ release into the myoplasm which then binds to the contractile proteins resulting in sarcomeric shortening (muscle contraction). ECC is terminated when Ca2+ is actively pumped back into the sarcoplasmic reticulum by the sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA) Ca2+ pumps. SRP-35 is a retinol dehydrogenase converting retinol to all trans-retinaldehyde, SRP-27 (TRIC-A) oligomerizes on the terminal cisternae (TC) and longitudinal sarcoplasmic reticulum membrane where it is thought to be involved in the transport of K+ ions during Ca2+ release to maintain a neutral electrochemical gradient across the sarcoplasmic reticulum membrane.
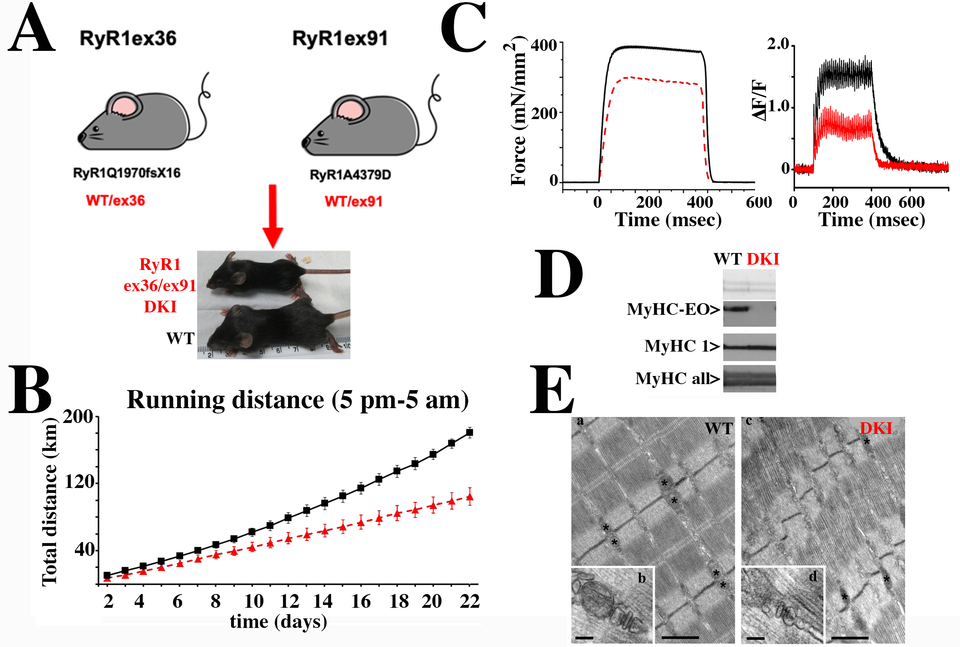
Fig. 2: Characterization of the DKI mouse model, knocked in for recessive RYR1 mutations. A. Double Knock In (DKI) mice were obtained by crossing heterozygous mice carrying a WT allele plus the frameshift mutation RyR1Q1970fsX16 with heterozygous mice carrying a WT allele plus the missense mutation RyR1A4329D. DKI mice (red) were on average 20% smaller than WT littermates (black). B. Muscle force assessed using the voluntary running wheel shows that DKI mice (red) run on average 50% less than their WT littermates (black). C. Ex vivo tetanic force stimulation of externsor digitorum longus muscles shows that muscles isolated from DKI mice (red) develop significantly less force than muscles from WT littermates (black)(left panel); the calcium transients elicited by electrical field stimulation at 150 Hz is also significantly reduced in EDL fibers from the DKI mice (right panel). D. EOMs from DKI mice show altered expression and content of the extra-ocular (EO) myosin heavy chain (MyHC) isoform. Top panel, membrane stained with Ponceau Red; central panel, blots stained with a monoclonal Ab specific for MyHC-EOM (top lanes) and a monoclonal Ab specific for MyHC1 (bottom lanes); bottom panel, blot stained with a monoclonal Ab recognizing all MyHC. E. Ultrastructure of EDL from WT and DKI mice. (a) In adult WT EDL fibers mitochondria are usually placed at the I band in proximity of Z lines (asterisks), next to CRUs. CRUs are mostly in the form of triads: two SR vesicles closely opposed to a central T-tubule (b). (c) In EDL fibers from DKI mice, mitochondria are less abundant and CRUs are often found in the form of dyads (d). Scale bars: a and c, 1 μm b and d, 0.1 μm.